Call +91-9823049121 / 7030024643
Mon - Sat: 9:00am - 9:00pm (IST) Sun: Closed
Call +91-9823049121 / 7030024643
Mon - Sat: 9:00am - 9:00pm (IST) Sun: Closed
This case involves a couple with a history of recurrent unfavorable obstetric outcomes, prompting them to seek genetic testing. The 33-year-old female, undergoing treatment for a prior hormonal imbalance, presented with ovarian hyperstimulation and a history of two previous abortions before 15 weeks of gestation. Initial microarray analysis on the product of conception of the first fetus yielded normal results, and no familial genetic abnormalities were identified.
Due to the absence of a family history related to genetic issues, the couple underwent whole exome sequencing (WES) to explore potential hereditary factors contributing to their recurrent pregnancy losses. WES revealed a homozygous polymorphism in the F5 gene, specifically a single nucleotide variant c.1601A>G[p. Gln534Arg].
This F5 gene mutation is associated with Thrombophilia 2, notably Factor V Leiden thrombophilia, exhibiting an autosomal dominant inheritance pattern with variable penetrance. Certain conditions of the F5 gene mutation are also linked to autosomal recessive conditions. Factor V Leiden thrombophilia is characterized by a blood clotting disorder, manifesting as a reduced anticoagulant response to activated protein C, potentially leading to thrombosis.
Numerous studies have confirmed a significant correlation between the identified F5 gene polymorphism and recurrent pregnancy loss. Women with this mutation are found to be at anincreased risk, being two to three times more likely to experience multiple miscarriages or pregnancy losses during the second or third trimester. Both partners in this case carry the F5 gene mutation, increasing the likelihood of passing it on to their offspring.
Although the couple has not exhibited any symptoms related to the F5 gene mutation, this could be a factor in their pregnancy challenges. The patient received counseling after the genetic test to understand the implications of the result for future pregnancies. With this information, they can now make informed decisions and a doctor can plan for proper management for their next pregnancy, addressing the root cause of their challenging obstetric history.
A 30-year-old female presented to the hospital after experiencing two spontaneous abortions, with no reported family history of similar occurrences. The reason for both early pregnancy losses was the loss of fetal heartbeat. Microarray analysis of the products of conception (POC) revealed a gain in chromosome 3, suggestive of trisomy 3. Concerned about the potential recurrence of trisomy in future pregnancies, the couple was subjected to karyotype analysis, which was normal. However, further examination identified a polymorphic variant in chromosome 22 in the female where the length of the short arm of chromosome is increased, this is previously associated with infertility and recurrent abortion.
Chromosomal polymorphic variations are usually considered normal findings; this occurs in 2–5% of the general population. Notably, the incidence rises to approximately 10–15% in individuals experiencing recurrent miscarriages compared to the general population.
These variations are predominantly manifested in heterochromatic regions, which are non-coding tandem DNA repeats. Importantly, variations in these regions do not result in noticeable phenotypic differences.
While the recurrence risk for trisomies in the next pregnancy is generally 1% in the absence of balanced translocation in parents, however, in this case, the presence of a polymorphic variation leads to complexity, making precise interpretation challenging.Considering the detected polymorphic variant and the recurrent miscarriage history, recent literature shows dataregarding potential advantages through assisted reproductive techniques, notably in vitro fertilization (IVF). IVF offers the prospect of selecting embryos with normal chromosomes, thereby reducing the likelihood of chromosomal abnormalities in subsequent pregnancies for this patient.
A 22-year-old female with a previous history of pregnancy loss in the 13th week of pregnancy presented for preconception screening. There is no family history of any genetic condition. Her recent CBC report was suggestive of lower MCV(mean corpuscular volume)and MCH (Mean corpuscular hemoglobin) levels, hemoglobin values were 7-8g/dl, and the HPLC report showed positive results for thalassemia.
Based on her previous history and clinical condition she was referred for a genetic test to confirm the thalassemia trait, this test revealed that she was a carrier forHbE-β-thalassemia variation [HbE+codon41/42(-CTTT)] thus confirming the cause of the spontaneous abortion. The βE allele is mildly thalassemia as this mutation gives rise to the HbE trait, also activates a cryptic splice site, and when inherited together with β° thalassemia, results in a marked deficiency of β chain production. The clinical and hematological changes of HbE/β thalassemia are variable, ranging from severe anemia and transfusion dependency to thalassemia intermedia. Such cases usually lead to anemia (hemoglobin values range from 4–9 g/dl) and splenomegaly.
Because the patient is affected by this condition, and if her partner is also a carrier for this condition, there is a chance that their children will develop the condition; therefore, a genetic test was performed on her husband to see if he is a carrier for this condition; however, the result revealed that he did not carry any mutation for thalassemia; thus, the risk of their children being affected by this condition is reduced because thalassemia is an autosomal recessive condition. The offspring will only be carriers of this disease. This knowledge about her thalassemia trait will help her plan and manage her future pregnancies effectively.
A 25-year-old pregnant woman reported a sudden eyesight issue, complaining of mist or fog covering one eye's vision. She was concerned since her maternal side of the family had a history of vision impairment, and she was also pregnant. Her mother is said to have muscular weakness, tremors, and mobility issues. Based on the patient's illness course and family history, a mitochondrial condition of optic neuropathy was suspected. As a result, she was referred to a mitochondrial disorders screening panel for an accurate diagnosis. The genetic test indicated that she carried the MT-ND6: mitochondrial encoded NADH dehydrogenase- 6 gene variant m.14484T>C, which is linked to Leber optic neuropathy (LHON).
In young adults, Leber hereditary optic neuropathy (LHON) often manifests as bilateral painless subacute blurred vision. Fundal abnormalities such as parapapillary telangiectasia vessels and varying degrees of retinal nerve fiber layer edema have previously been described in the presymptomatic period; they can alter over time. Visual loss is bilateral at the outset in 25%-50% of people. The most characteristic feature is an enlarging central or centrocecal scotoma, and as the field defect increases in size and density, visual acuity deteriorates to the level of counting fingers or worse.
Cascade testing for this mutation in the family was suggested to prevent or manage the condition. This variation was also found in the patient's younger sibling, confirming the disease's maternal inheritance. A discovered mutation does not always result in visual loss, but her brother is at a 50% lifetime risk of developing the disorder. Because the patient's mother had LHON-related symptoms, younger members of her maternal side family were referred for variant screening. Prenatal testing was not suggested for the condition of this patient because mtDNA mutation in amnio/cvs samples makes interpretation challenging because of the varied penetrance and onset. With a significant LHON-causing mtDNA pathogenic mutation, about 50% of males and 90% of females do not develop blindness. Based on the genetic variation identified within this family, males are 47% more likely than females to develop the symptoms of the disease.
This genetic screening helped the patient and other family members to identify the mitochondrial disease running in the family
A non-consanguineous couple seeking preconception counseling had a history of abortions due to fetal abnormalities. The 32-year-old female partner was found to have diabetes, hypothyroidism, and hypertension. She had a history of abortions between 12 and 20 weeks gestation, with abnormal ultrasound findings such as Acrania, unossifiedcalvarium, cranial vault, and bilateral talipesdeformity.
The couple was advised to undergo whole exome sequencing to rule out any genetic conditions that might be causing congenital abnormalities during pregnancy. The female partner report indicated a unique heterozygous mutation in the STAG2 gene, which is linked to Mulegama-klein Syndrome (MKMS). This is a rare X-linked disorder caused by a heterozygous or hemizygous variant in the STAG2 gene. MKMS is characterized by a variety of clinical symptoms, including developmental delay, intellectual impairment, craniofacial abnormalities, and brain malformations. Global developmental delay, increasing microcephaly, speech and language delay, and poor overall growth. The affected individuals are reported to have microtia with hearing loss, attention deficit-hyperactivity disorder, failure to thrive, short stature, foot polydactyly, pes planus, and unsteady gait with lower limb hypotonia.

Based on the findings of this genetic investigation, we may conclude that the mutation detected in the female is a likely cause of fetal genetic abnormalities. Males are more likely to be affected by this condition since it is an X-linked disease, which means that the genetic characteristic is passed down from mother to fetus by mutations in an X-chromosome gene. The abnormalities discovered in the pathology report of a G2 male fetus correlate with the symptoms of this MKMS disease. The whole exome sequencing identified the possible cause of their poor obstetric history and based on these findings the couple can make an informed decision for their next conception with the necessary genetic investigation prenatally as suggested by the doctor.
A non-consanguineous couple presented with a concern that their 7-year-old son had congenital vision loss, as well as their second child, who was 4 years old and suspected of having vision loss. This couple is planning their next pregnancy but is concerned about their children's eyesight impairment and risk for next pregnancy, so they were referred for genetic counselling.
Following counselling, the affected son i.e. the family's index case was referred for an exome analysis, which indicated that he carried a pathogenic heterozygous mutation in the LRP5 gene, which is associated with Familial Exudative Vitreoretinopathy. This is a genetic condition that can lead to gradual visual loss, the retina is affected by this disorder. It inhibits blood vessels from developing along the retina's borders, reducing blood flow to this area. LRP5 genes provide instructions for making proteins that participate in a chemical signalling pathway that affects the way cells and tissues develop.The indications and symptoms of familial exudative vitreoretinopathy vary greatly, even within the same family. Many people with retinal defects never have any visual difficulties. In others, a decrease in the retina's blood flow leads the retina to fold, rip, or detach from the back of the eye. This retinal injury can result in vision loss and blindness. The incidence of familial exudative vitreoretinopathy is unclear. It appears to be uncommon, yet afflicted persons with normal vision may never seek medical attention.
Later, this similar variant in the LRP5 gene was tested in the couple and second child, revealing that the mother of the index case carried a similar mutation but had no symptoms associated with the condition, while the symptomatic daughter (second child) carried the same heterozygous mutation. Despite the fact that the illness is inherited in an autosomal dominant pattern, because of its variable penetrance in the family, only the children are impacted, making future pregnancy risk prediction challenging. As a result, she was advised to undergo prenatal testing to see whether the foetus is impacted by the same mutation and to make a well-informed decision.
This case is an example of how genetic screening has been helpful in identifying inheritable condition in the family, the onset of which can occur at any age, and in planning better treatment for affected children as well as making a well-informed choice for the next pregnancy.
This is a case of a non-consanguineous couple presented with a congenital genetic abnormality in first pregnancy. The baby was found to have severe IUGR and foetal distress after delivery, initially suspected with Microcephaly, MOPD (Microcephaly Osteopdysplastic Dwarphism), Seckel syndrome, Osteoplastic, and Dwarphism. Baby's response to growth hormone therapy was minimal. Later she was diagnosed with MOPD.Baby was subjected to whole exome analysis, which showed that she was homozygous for a pathogenic mutation in the PCNT gene, at position Val3179Glyfs*36 (c.9535dup). This gene is associated with MOPD, characterized by intrauterine growth retardation, severe proportionate short stature, and microcephaly. Hence the diagnosis in this baby was confirmed for this condition.
Later, this couple underwent preconception screening, which revealed that they were both carriers for the same variant in the PCNT gene associated with MOPD. As this is an autosomal recessivecondition, the couple has not developed any symptoms. In such cases, in her future pregnancy, there is a 25% probabilityfor the baby being affected with this condition and 50% chance of being a carrier and 25% chance of being normal.
They planned for the next pregnancy after receiving appropriate genetic counselling and understanding all of their possible pregnancy outcomes. In the 12th weeks of conception prenatal genetic testing was performed to detect the presence of the PCNT gene variation in thechorionic villus sample by sanger sequencing and also microarray analysis was performed to rule out the possibility of chromosomal abnormality in the developing fetus. This CVS analysis detected a heterozygous variant for the PCNT gene,one copy of mutated gene is present which represents a carrier statefor this baby without developing any signs and symptoms and also the microarray analysis was normal. Because of no significant prenatal findings, they continued with this pregnancy.She gave birth to a healthy baby with no skeletal deformities or dysmorphism. There were no significant findings in this baby's newborn genetic screening.
This case is an example of how genetic screening at various stages of conception helped the couple to make an informed decision.
A non-consanguineous couple presented withbad obstetric history with multiple abortions suspected with a high risk pregnancy for trisomy 21 in the second trimester. The couple karyotyping was normal. Female’s blood reports were suggestive of Vitamin B12 deficiency, ANA –positive. She was referred for MTHFR gene mutation analysis, MTHFR is responsible for the breakdown of folic acid, which creates folate. Some health conditions and disorders can result without enough folate, or with a malfunctioning MTHFR gene. This can cause a reduced enzyme activity leading to elevated homocysteine levels and these elevated levels can lead to cardiovascular disease, deep venous thrombosis and preeclampsia andother pregnancy complications.
She was detected with homozygous mutation in the MTHFR gene c.1298 A>C (lesser reported variant), suggestive of low enzyme activity. High homocysteine levels, especially with low folic acid levels, can lead to pregnancy complications like higher risk for miscarriages, preeclampsia or a baby born with birth defects, such as spina bifida. As per literature homozygosity for MTHFR A1298C is (early-late) risk factor for Recurrent Pregnancy Loss (RPL)(N Mtiraoui et.al., 2006). While the exact cause of RPL remains undetermined in most of the cases, genetic predisposition to venous thrombosis (Finan et al. 2002) and elevation in total homocysteine levels (hyperhomocysteinemia) have been described as playing a role in the pathogenesis of RPL. This test will help to rule out these possible complications associated with malfunctioning of MTHFR gene by including proper amount of folic acid in diet each day from fortified foods and vitamins/supplements before and during pregnancy
MTHFR gene mutation analysis can help in guiding proper folic acid consumption for a pregnant woman.
Anon-consanguineous couple presented to doctor with a history of two female neonatal deaths and required guidance before they plan for the next pregnancy. Both the babies had developed acute intestinal obstruction at the very initial stage of life (within 1 month), suspected with meconium ileus. The karyotype analysis for the couple and second female child were normal. Due to all these findings the couple were referred for genetic counselling, they (couple) were recommended to go for whole exome sequencing before they plan for their next pregnancy.
This genetic evaluation showed that the couple did not carry any significant gene mutation in association with meconium ileus. But they both were a carrier for a pathogenic heterozygous variant in the HFE gene known to cause hemochromatosis. This gene provides instructions for producing a protein that is located on the surface of cells, primarily liver and intestinal cells and is also found on some immune system cells.Main function of this geneis to regulate the production of a protein called hepcidin whichis produced by the liver, and it determines how much iron is absorbed from the diet and released from storage sites in the body. Hereditary hemochromatosis with decreased hepcidin expression leads to iron-overload disorder that causes increased intestinal iron absorption and accumulation of excessive iron in other organsas well \ this can result in morbidity and mortality if not treated/ in case of severe condition.Few research papers have mentioned that; in rare case scenario excessive iron deposition can lead to intestinal obstructions, hence this condition might be the cause for the previous neonatal death (Bharadhwaj Ravindhran et.al.,2018) (Swetha Parvataneni et.al.,2020)
As the coupleare the carrier for this condition; their children have a higherrisk of being affected by this condition. This information helped the couple take an informed decision before planning for next pregnancy, which would have otherwise gone undetected if this test was not performed.
A couple with consanguineous marriage was presented with a bad obstetrics history. They had experienced five recurrent miscarriages; all of them before 8th month of gestation. Clinical findings during the conception were indicative of conditions like polyhydramnios, fetal ascites, recurrent infections, fetal hydrops, bilateral hypo plastic lungs with fetal hepatomegaly and fetal tachycardia. The karyotyping analysis of the couple were not suggestive of any significant findings in association with pregnancy loss; showing a normal 46XY karyotype for male and female as 46XX 21 psi+karyotype. As there were no findings with any other tests, the couple underwent exome testing. This test revealed that both of them werecarrier for a mutation in the MVK gene which is known to cause Mevalonate kinase deficiency (Mevalonic aciduria).

This is a metabolic auto inflammatory syndrome caused by mutations in the MVK gene, mevalonate kinase, the key enzyme in the non- sterol isoprenoid biosynthesis pathway. Two phenotypes of mevalonate kinase deficiency are known based on the level of enzymatic deficiency, mevalonic aciduria and hyperimmunoglobulinemia D syndrome. Mevalonic aciduria is characterized by dysmorphology, psychomotor retardation, progressive cerebellar ataxia, and recurrent febrile crises (similar to those observed in hyperimmunoglobulinemia D and to periodic fever syndrome), usually manifesting in early infancy, accompanied by hepatosplenomegaly, lymphadenopathy, arthralgia and skin rash. Some severe cases of mevalonic aciduria are also characterized by hydrops fetalis, foetal ascites, hydrocele, hepatosplenomegaly, intrauterine growth restriction and polyhydramnios.
The novel variant detected in this case has not been reported before as pathogenic in association with mevalonic aciduria conditions, hence this variant is classified as uncertain significance. Because there is a significant correlation between these conditions and the clinical findings observed during recurrent pregnancy loss in this test report, it is suspected that this gene is responsible for the conditions observed in the developing foetus that leads to pregnancy loss. This is an autosomal recessive condition and as the couple is a carrier for the same variant in the MVK gene, there is a high probability that their baby would be affected by this condition. Hence, in this case a whole exome analysis has helped the couple to make an informed discussion about their future conception.
This is a case of a 37 years old primi who was presented with no obvious clinical findings for any fetal abnormality except for risk associated with her advanced maternal age. The ultrasound performed in the 12th week of gestation was observed to be normal with adequate fetal growth and liquor quantity.The estimated risk for trisomy 21 based on her maternal age and fetal nuchal translucency thickness for crown-rump length was 1 in 210.
Due to her advanced maternal age she was further referred for Non-Invasive prenatal screening (NIPS) test in her 13th week of gestation to detect if the fetus has a risk for developing any chromosomal aneuploidiesusing cell free fetal DNA form the maternal blood without posing any risk to the fetus. This chromosomal evaluation predicted a high risk for trisomy21. Trisomy 21 is the most prevalent genetic disease worldwide and the common genetic cause of intellectual disabilities appearing in about 1 in 400-1500 new-borns.This finding from NIPS test was confirmed using invasive prenatal testing for which the patients underwent an amniocentesis procedure in her 17th week of gestation, this amnio sample was further studied by Fluorescent in Situ hybridization (FISH) method. This specific probe based analysis was also positive for trisomy 21.

Due tohigh risk pregnancy (trisomy 21) and possibility of future pregnancy related complications, this pregnancy was discontinued. The skin scarping from the abortus fetus was further tested using chromosomal microarray analysis. This test showed that there was a gain of an additional copy of chromosome 21 at position p11.2 to q22.3 representing a complete gain of chromosome 21; suggestive of Trisomy 21-Down’s syndrome. This result correlates with the risk estimated in both invasive and non-invasive prenatal testing.
This case shows that the sensitivity of NIPS for detecting chromosomal aneuploidies is very high, this is an ideal test to be performed at the very initial stage of pregnancy to estimate the risk for numerical chromosomal aneuploidy in the fetus especially for trisomy 21,18 and 13. This test can help pick up these condition before any invasive tests can even be performed.
This is a case of a 6 years old child with clinical presentations like delayed milestones, hyperactive nature, cardiac myomas and patches on the skin hence suspected with tuberous sclerosis. There is no known history of this disorder in their family. Tuberous sclerosis is an uncommon genetic disorder affecting 1 in 6,000 to 10,000 people. It causes noncancerous tumors to develop in many parts of the body and has a varied age of onset; which can develop in infancy/ childhood/late adulthood or sometimes even left undiagnosed. The symptoms also vary widely, depending on where the tumor develops and how severely a person is affected. It is generally characterized by abnormalities of the skin, brain, intellectual disability, developmental delay, psychiatric illness, kidney and heart issues etc. Central nervous system tumors are the leading cause of morbidity and mortality; renal disease is the second leading cause of early death. Hence to confirm the exact diagnosis in this case, the child was subjected for a whole exome sequencing which would cover all the genes associated with the phenotypes.
This genetic test revealed that she harbors a heterozygous pathogenic variant in TSC2 gene present in exon4; with variant nomenclature as c.328C>T leading to a protein change of p. Gln110Ter. This variant is predicted to cause loss of normal protein function which leads to this disease condition, thus confirming her diagnosis for Tuberous sclerosis. This is an autosomal dominant condition where only one copy of the mutated gene is required to develop the condition because of which she has started developing the signs and symptoms for this disease condition at a very early age.
Due to the varied age of onset for this disorder, patient’s asymptomatic parents were also subjected for the same variant analysis by sanger sequencing. This test was performed to know if they are also the carrier for the same mutation which might increase their risk of developing this disorder in the future. Genetic evaluation confirmed that both the parent were non-carriers for this heterozygous variant which means they are not genetically predisposed with the risk of being affected. Hence, concluding that the affected child has developed a sporadic mutation for Tuberous Sclerosis and not inherited it from either of her parents.
This genetic test has helped the family to know that the child is affected with Tuberous sclerosis and plan the medical surveillance accordingly. Although there is no cure for this disorder, treatments are available to manage the developing symptoms.

This is a case of a 5 years old childwith high grade fever and persistent cough for 4 days, presenting additional symptoms like weakness and loss of appetite. In spite of being on anti- viralmedication for over4 days there was no improvement seen in her health and was suspected with respiratory tract infection.Asvariety of viruses and some bacteria can cause infections of the respiratory tract, she was referred for a RT-PCR based respiratory pathogen panel testing for specific detection of the type of infection causing thesymptoms of respiratory illness.
This panel screens for 31 respiratory pathogens to determine whether thereis a respiratory infection due to certain bacteria or viruses; that can help guide management of the existing infection which could be the cause of recurrent fever and cough in this case.
The Naso-oropharyngeal swab of the child was tested.This test detected a bacterial infection of Haemophilus influenza and a viral infection of Parainfluenza virus 1, indicative of a co-infection. These pathogenscould be the reason forvisible signs and symptoms in the child. Based on this report;doctor had started antibiotics for her immediately, which showed faster recovery from fever and other symptoms.
Respiratory pathogen testing helped the doctor to modify the treatment plan for the child based on the specific pathogen detected.
This is a case of a couple with a history of missed abortion. The microarray analysis of the abortus sample was performed using the fetal skin scraping which detected a gain of 1600 kbp on X chromosome- arr [GRCh 37] Xp22.31(6486490_8086425) x 3. Due to this finding the couple were subjected to a microarray analysis to know if the detected copy number gain in the abortus sample was spontaneous or inherited from either of the parent. This chromosomal microarray analysis showed that the female carried two copy number variants representing arr [GRCh 37] Xp22.31(6486490_8116696) x 3 which is a gain of 1630 kbp and a loss of 210 kbp on the Xq13.3 region. This 1630kbp gain is similar to the gain identified in the abortus sample. The Xp22.31 segment of the human X chromosome reported to be highly instable region with frequent rearrangement. Hence, this has a varied penetrance and is reported in healthy as well as symptomatic individuals.
This region includes the OMIM genes namely PUDP, STS, VCX and PNPLA4 and has been reported to be associated with conditions like X-linked ichthyosis, hypotonia, cardiomyopathy, familial hypertrophic 7, atrial septal defects, intellectual disability, autism and other disorders. The prevalence of this CNV associated abnormality is around 0.23% in males and 0.38% in females. This is known to cause a major causative effect if there is a gene mutation along with this chromosomal gain. This study shows that there is a possibility of inheriting the copy number variant in the fetus which could be possible reason for developing a pathological condition which subsequently led to the abortion. This case is an example of how chromosomal microarray analysis helps in detecting the possible inheritance of a CNV (micro duplications/microdeletions) which can help the couple to make an informed decision for the next pregnancy.
A case of a consanguineous couple with a bad obstetric history of 3 recurrent miscarriages and one neonatal death due tounexplained reasons (no proper diagnosis). The clinical findings in infant werehepatosplenomegaly, severe weakness,poor feeding, low weight gains and was suspected to havemetabolic disorders. After this; the couple was recommended to undergo a whole exomescreening test to understand if any genetic condition is being inherited to theiroffspring, but they refused to get tested. After 2 years; she delivered a healthy female child (5thconception) with no clinical diagnosis like her previous baby, but considering her obstetrics history and one unexplained neonatal death, her baby girl was referred for a new-born genetic screening test.
This screening test detected a pathogenic variant in the MPV17 gene known to cause Mitochondrial DNA depletion type 6syndrome.
MPV17-related hepatocerebral mitochondrial DNA depletion syndrome is an inherited disorder that can cause liver disease and neurological problems. The signs and symptoms of this condition include vomiting, diarrhoea, lactic acidosis, hypoglycaemia, hepatosplenomegaly (enlarged liver)and an inability to grow or gain weight at the expected rate.This condition is inherited in a mitochondrial inheritance pattern.
This condition purely follows a maternal inheritance pattern, male and females are equally affected but even an affected male cannot inherit this condition to his next generation. The symptom onset age and its severity may vary from person to person.
This screening report suggests that the new born baby is having an increased risk of developing the mitochondrial condition and henceadvised to keep her under medical surveillance.Theroutine medical check-up shows that the baby has started developing a slight liver enlargement and flattering growth (poor weight gain),these clinical findings correlated with the mitochondrial condition identified which has helped in proper management for the baby.This case shows how new born genetic screening helps with on time medical intervention by picking up the condition while the baby is asymptomatic.
An asymptomatic couple had two completely deaf daughters, their genetic screening test revealed that both the sisters were homozygous for a pathogenic mutation in the GJB2 gene (W24X, c.71 G>A). This gene provides instructions for making a protein called gap junction beta 2, more commonly known as connexin 26. This protein is found in the cells throughout the body, including the inner ear. Because of its presence in the inner ear, especially in the snail-shaped structure called the cochlea, this is an important gene associated with non-syndromic hearing loss.
The c.71G>A (p. W24X) mutation in GJB2 gene is a nonsense mutation consisting of a G-to-A transition at position 71, resulting in a trp24-to-ter substitution (24 amino acids instead of 226 amino acid). This mutation is associated with autosomal recessive non-syndromic sensorineural hearing loss where two mutated copies of the genes are required to develop the symptoms for this disorder. Genetic study for the parents revealed that they were the heterozygous carrier for the same mutation, butboth of themwere asymptomatic as they carried only one mutated copy of the gene.
Their daughter who was homozygous for GJB2 gene mutation got married to a person with similar conditionand also had a similar family history, so they went through a preconception genetic analysis; by screening the GJB2 gene to check if both the partner carry similar mutation in GJB2 gene.

His genetic test revealed that he wascompound heterozygote for the GJB2 gene (W24X, c.71G>A) and (R32L, c.95G>T). In such scenario there is a very high chance that their baby (male or female)will inherit the mutation from them and develop the condition. So, this couple opted to conceive through in-vitro fertilization (IVF) techniqueusing a donoras per the medical geneticist’s recommendation. She delivered a healthy female baby, and the baby was also subjected for genetic screening to know if she carried the same mutation like her parents.
Her genetic test revealed that she was just a carrier for only one copy of the mutation in GJB2 gene (W24X, c.71G>A), this being an autosomal recessive disorder the baby will remain asymptomatic for this condition without developing any hearing impairment. Thus reducing her risk for being affected by a homozygous mutation like her parents.
A 44 years old female(patient) with a clinical history of three recurrent abortions conceived normally and delivered a healthy baby girl (age=14 years). Her daughter is reportedly healthy without any medical issues so far. Post-delivery(after 7 years) she naturally conceived 3 times, which was not successful.
Patient and her partner have no known family history of any genetic disorder. But looking at the bad obstetrics history of the patient ,the couple were referred for genetic carrier screeningto rule out the possibility of being affected with some recessive/dominant genetic disorders.
The genetic analysis reveals a mutation in the couple in the HBA2 gene with copy number variation (CNV) – loss which is associated with alpha thalassemia. Patient is a compound heterozygote (when both alleles of a gene posses’ mutations, but the mutations are different, these mutations are called compound heterozygous) for the CNV- loss mutation and her husband is a silent carrier(inherited 3 normal alpha-globin genes (-α/αα) are referred clinically as silent carriers) for the mutation in the same gene.
This detected mutation is pathogenic and pregnancy in thalassemia patients can be complicated with miscarriages, fetal loss, preterm delivery, IUGR, and thrombosis. The couple being in heterozygous carrier state; there is a 25% chance of both passing on the defective copies to the offspring which can be associated with the chance of abortions.
A 31 years’ female with a family history of cancer diagnosed with triple negative breast cancer (Stage I) at the age of 29 years, indicative of lump on the right breast with highly suspicious malignant cells. Patient underwent a lump removal along with Latissimus Dorsi flap reconstruction therapy (cosmetic surgery) and her biopsy report were indicative of medullary carcinoma and negative for sentinel nodes. Post-surgery she had received chemo and radiation therapy.
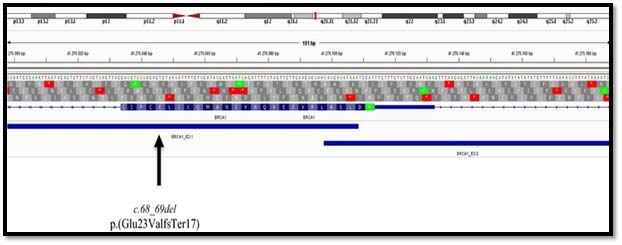
She was referred for BRCAI/II mutation considering her early age of diagnosis and a family history. The genetic mutation analysis detected a pathogenic variation as reported in ENIGMA consortium and Clinvar database on chromosome 17- c.68-69delAG (p. Glu23ValfsTer17). This mutation causes 2 base pair deletion in exon-2 causing frameshift in the amino acid sequence beginning at the 23rd position of the resultant protein; this introduces a downstream stop codon at 40th amino acid, which leads to truncation of the protein at 40aa from its original length of 1885aa.
As BRCAI is a tumor suppressor gene, this reported truncation/mutation in protein hampers its function subsequently leading to overgrowth of cells uncontrollably; resulting in a tumor formation. As per data, the identified mutation has been associated with Familial breast cancer cases and hereditary breast and ovarian cancer syndrome.
From this germline mutation analysis, we can conclude that she might have inherited one abnormal copy of BRCAI gene from her paternal side of the family which made her highly susceptible for developing breast cancer at an early age; this can be confirmed by targeted mutation analysis of this identified mutation in the proband.
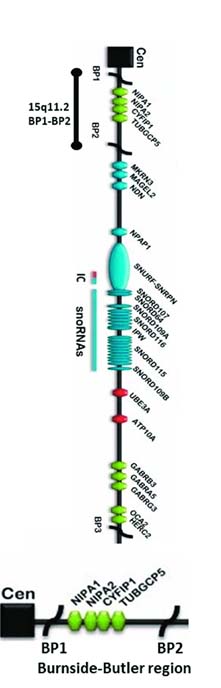
A 22 weeks’ pregnancy presented hypoplastic nasal bone in congenital anomaly scan after termination further microarray analysis on the POC (Product of Conception) sample showed that, the fetus had a 15q11.2 microdeletions. Subsequently the couple was referred for CMA analysis for which the mother was reported as normal but father was detected with the same 15q11.2 microdeletions, this being an autosomal dominant condition; it concludes that the fetus had inherited this deleterious mutation from its father. Even though the mutation has been detected in the patient (father), he has no visibility associated phenotypic characters.
The insilico analysis anticipates that; this is a rare chromosomal abnormality known as Burnside Butler syndrome/15q11.2 microdeletion syndrome. This microdeletion involves four genes (i.e., TUBGCP5, CYFIP1, NIPA1, NIPA2) with a prevalence ranging from 0.57%–1.27% The four deleted genes in this syndrome are mainly associated with the neurodevelopmental disorders with change in brain morphology, cognitive impairment, language and/or motor delay, autism, behavioural problems, poor coordination, ataxia, and congenital anomalies.
A detailed study of the family history revealed that the patients’s (father’s) cousin brother has an intellectual disability, this character has also been reported to be associated with the same microdeletion syndrome. As observed in this case scenario; various literatures have also reported that this particular chromosomal abnormality has a variable penetrance for each case detected due to which few affected individual are healthy and does not develop any phenotypic characters.

A couple with normal history and non - consanguineous marriage presented with 3 consecutive neonatal deaths due combined immunodeficiency along with skin eruption which was suspected to have congenital ichthyosiform erythroderma.
Congenital ichthyosiform erythroderma (CIE) is characterized by fine, whitish scales on a background of erythematous skin over the whole body. CIE is an autosomal recessive genetic disorder. This means a child must inherit a defective pair of genes (one from each parent) to show the symptoms.
Parents who are carriers of the defective genes show no symptoms but their children have a 25% chance of having CIE.
We performed whole exome sequencing (WES) of the couple for detection of variants and their functional significance using several online prediction tools. The result revealed that the couple carried a heterozygous variant in RAG2 located in exon 3 of the gene (NM_00536.4, c.137 T > C [p.Phe46Ser]). The Insilco analysis anticipated it to cause severe combined immunodeficiency and it is inherited in autosomal recessive manner.